Introducción
Los sarcomas son tumores relativamente poco comunes representando, aproximadamente, el 1% de todos los cánceres de adultos . Constituyen un grupo heterogéneo de tumores de origen celular mesenquimático con distintos distribución de edad, lugar de presentación, comportamiento biológico natural y pronóstico. Hay más de 50 subtipos divididos en dos grandes categorías: sarcomas de partes blandas (SPB) y sarcomas óseos (SO).
En 2010 alrededor de 3.300 personas fueron diagnosticadas de SPB en el Reino Unido, con unos 90 casos en niños menores de 15 años. En el grupo de edad entre 17 y 25 años se registraron alrededor de 80 casos . La National Cancer Intelligence Network informa que la incidencia de los SPB es de aproximadamente 45/1.000.000 de habitantes al año .
Los sarcomas óseos son más raros, con una incidencia cercana al quinto de los SPB; en 2011 se registraron 559 nuevos casos . Sin embargo, representan una proporción significativa de incidencia de cáncer en los jóvenes menores de 20 años. Los SPB pueden aparecer en cualquier parte del cuerpo, a cualquier edad y con mayor frecuencia en adultos de mediana edad y adultos mayores; sin embargo, proporcionalmente, dentro de las neoplasias pediátricas, son relativamente comunes, suponiendo el 7-10% de todos los cánceres infantiles.
Son una importante causa de muerte en el grupo de edad 14-29 años. Aproximadamente la mitad de todos los pacientes con SPB con tumores intermedios o de alto grado desarrollan enfermedad metastásica que requiere tratamiento sistémico . La supervivencia general es de aproximadamente 55% a los 5 años. La etiología de la mayor parte de los SPB es desconocida. Entre los factores de riesgo de los SPB infantiles se han propuesto:
- Factores de riesgo constitucionales: Síndrome de Li-Fraumeni, neurofi bromatosis tipo 113, el síndrome de Gardner e inmunodefi ciencias.
- Factores de riesgo medioambientales:
- Directas sobre el feto: radiaciones ionizantes en el útero y otros genotóxicos.
- Indirectas: exposiciones ocupacionales a herbicidas, dioxinas y cloruro de vinilo, consumo de drogas (tabaco, marihuana y cocaína) durante el embarazo y bajo nivel socioeconómico de los progenitores.
En este artículo, a partir de una revisión de los aspectos clínicos y epidemiológicos de los tumores malignos de origen fibroso se propone una guía que facilite el diagnóstico clínico al profesional de atención primaria, en general, y al podólogo de estos tumores que afectan al pie y a la extremidad inferior.
DESCRIPCIÓN DE LOS DE LOS TUMORES MALIGNOS DE ORIGEN FIBROSO
Entre los tumores malignos de origen fibroso con manifestación cutánea hemos incluido el fibrosarcoma (FS), el dermatofibrosarcoma protuberans (DFSP) y el histiocitoma fibroso maligno (HFM).
1. FIBROSARCOMA
La OMS define al FS como un tumor maligno que se caracteriza por la presencia de haces entrelazados de fibras de colágeno formadas por las células tumorales y por la ausencia de otros tipos de diferenciación histológica, tales como la formación de cartílago y hueso.
Es una entidad que se origina del tejido conectivo de sostén de la cavidad medular (el denominado fibrosarcoma central, endostal o medular) o, mucho menos frecuentemente, del periostio (fibrosarcoma perióstico) o de los tejidos blandos paraosteales. El FS, en un primer estadio, es similar en su aspecto al fibroma.
Puede alcanzar un tamaño importante hasta formar un gran tumor multilobulado, duro, renitente o seudofluctuante, de color rojo violáceo, telangiectásico, con tendencia a la ulceración; destruye los tejidos adyacentes desarrolla metástasis por vía hemática, con más frecuencia que la vía linfática. Se han descrito formas amelanóticas. Se han localizado FS cutáneo en la dermis y en el tejido subcutáneo de cualquier parte del cuerpo y de la cavidad oral, con preferencia por las extremidades. En el pie se han comunicado casos en el talón, en los dedos, en las plantas de los pies, diseminándose a lo largo de los tendones o fascias y con especial predilección por los nervios y los vasos sanguíneos. En cuanto a su localización, se acepta el predominio en extremidades (58-71%), frente a las localizaciones axiales en tronco (25 %), cabeza y cuello (17 %).
Como el fi brohistiocitoma maligno, el 30% de los FS son secundarios a la transformación maligna de lesiones benignas preexistentes (FS secundario), como displasia fibrosa, enfermedad de Paget ósea, infarto o quiste óseo y osteomielitis a radioterapia sobre hueso (tumor de células gigantes irradiado). También como consecuencia o evolución de un dermatofibrosarcoma protuberans. Clínicamente, se presenta entre la tercera y la sexta década (con una edad media de 59 años) pero se puede presentar a cualquier edad, incluso en los niños, habiéndose descrito formas congénitas.
En el FS óseo el diagnóstico radiológico muestra, a menudo, imagen osteolítica permeativa o moteada, con bordes mal delimitados y amplios con escasa o nula esclerosis reactiva y, generalmente, sin reacción perióstica. generalmente es de localización excéntrica en la metáfi sis y se extiende a epífisis o díafisis. Habitualmente hay una masa de tejidos blandos. La TAC demuestra que presenta una densidad similar al músculo. En ocasiones se ven áreas de menor densidad dentro del tumor que representan zonas de necrosis.
La gammagrafía, por su parte, muestra un área de incremento de captación, frecuentemente, en la periferia del tumor. Respecto a la etiología se acepta que el factor más importante en el desarrollo del carcinoma es la irritación crónica, ya sea mecánica, repetitiva o química. En este sentido, Walker, en 1941, propuso unas premisas que siguen teniendo plena validez en la actualidad. Son las siguientes:
- La lesión puede causar enfermedad.
- La lesión puede precipitar la enfermedad
- La lesión puede agravar la enfermedad.
- La enfermedad puede promover la lesión (“determinismo traumático” de Ewing)
- La lesión puede revelar enfermedad.
- La asociación puede ser pura coincidencia.
- En la debilidad post-traumática se pueden instalar diversas enfermedades y degeneraciones.
Es un tumor lentamente agresivo y produce metástasis en el 50% de los casos (principalmente en hueso, ganglios linfáticos y en pulmón) aún después de la cirugía radical. La supervivencia a los 10 años es del 28%, mejor para la localización periostio respecto de la central (40% frente al 20% de supervivencia a los 10 años).
Se han comunicado FS enmascarados por un hemangioma, un hemangioma ulcerado y alrededor de una osteomielitis crónica. Requiere diagnóstico diferencial con otras lesiones benignas y malignas con las que comparte un aspecto clínico similar en algunas fases de su evolución como los fi bromas, algunos adenomas, HFM, tumor de células gigantes, mieloma solitario, linfoma y sarcoma de Ewing, entre otros.

2. DERMATOFIBROSARCOMA PROTUBERANS
DFSP fue descrito por primera vez por Darier y Ferrand quienes denominaron a un grupo de tumores “dermatofi bromes progressifs et recidivants ou fi brosarcomes de la peau” (“dermatofi bromas progresivos y recidivantes de la piel”), en 1924. Su denominación actual se debe a Hoffmann, en 1925.
En 1962, Taylor y Helwig, en una revisión de 115 casos, describieron detalladamente las características histológicas del DFSP y lo catalogaron como un crecimiento fibroblástico que aparece como un sarcoma de bajo grado. El DFSP es un sarcoma de bajo grado de malignidad, con frecuentes recurrencias clínicas (alrededor del 60%) y raro potencial metastático (5% de los casos) pero con gran capacidad de destrucción local.
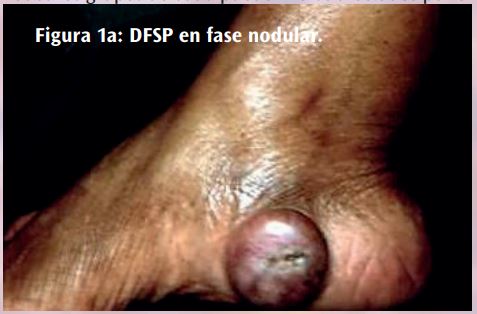
El DFSP suele ser de crecimiento lento, presentándose al principio como máculas o placas induradas, indoloras, pequeñas y del color de la piel, violáceas o marrón- rojizo. Después se vuelve protuberante y se convierte en una lesión multinodular. Los nódulos suelen ser de consistencia firme e irregular, adheridos a la piel pero libres en el plano profundo, y de color blanco-rojizo65 (Figura 1) .
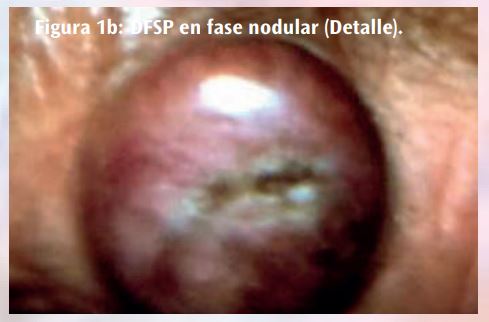

Todos los grupos de edad pueden verse afectados por el DFSP, incluidos los niños y los lactantes, pero es más común en la tercera a la quinta décadas de vida. Se presenta más comúnmente en la cintura torácica (entre el 50 y el 60% de los casos), las extremidades superiores, la cabeza y el cuello. Pero también se han descrito localizaciones acrales, en los pies incluida la fascia plantar, en los dedos de los pies e, incluso, unas formas pigmentadas que suponen entre el 1 y el 5% de los DFSP.
La tasa de incidencia anual es de, aproximadamente 5/1.000.000 de personas. Representa el 1% de todos los sarcomas de partes blandas. Según un estudio, la distribución por sexo es aproximadamente igual, con un ligero predominio femenino. El DFSP es de etiología desconocida aunque se han descrito formas congénitas y genéticas. También se han propuesto varios factores que pueden resultar desencadenantes, como un traumatismo.
El diagnóstico clínico del DFSP es difícil porque, además de ser un tumor muy raro, las características clínicas pueden se irrelevantes y su evolución suele ser lentamente progresiva. Sin embargo, el diagnóstico histopatológico es característico, con haces fusocelulares homogéneos celularmente, ordenados en verticilos o en rueda de carro. Invade la grasa en un patrón característico, rodeando individualmente los adipocitos para integrarlos en la masa tumoral.
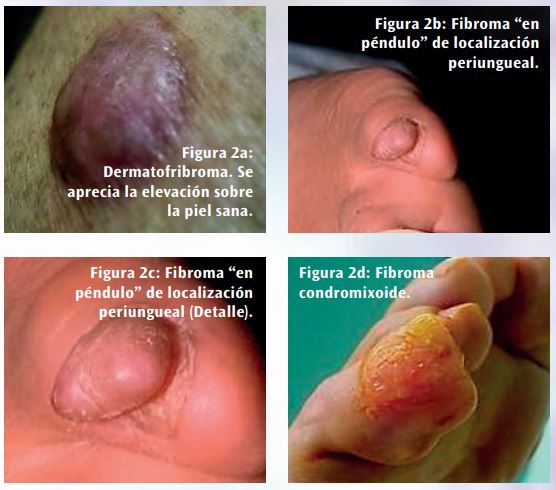
El componente superficial es similar de cerca al de un neurofibroma rodeando, en lugar de desplazar, los anejos cutáneos. La lesión es positiva para CD34, un marcador relativamente específico para diferenciarlo de otras proliferaciones fibroblásticas como el dermatofibroma. El diagnóstico diferencial del DFSP, clínico y anatomopatológico, hay que hacerlo con diversos tipos de tumores benignos de origen fibroso, muscular y lipocítico, como el dermatofibroma (Figura 2a), con varios tipos de fibromas (Figuras 2b, 2c, y 2d), especialmente los escleróticos, con el tumor de células gigantes, con el leiomioma y con el lipoma.
También con tumores malignos del mismo o similar origen como el leiomiosarcoma, el histiocitoma fibroso maligno, el fibrosarcoma y el liposarcoma. Como quiera que existen formas pigmentadas del DFSP, además de tener un patrón que puede semejar un tumor de origen nervioso, también deberá hacerse diagnóstico diferencial con nevus melanocíticos, con el neurofi broma pigmentado, con el swannoma melanótico y con el neurofi brosarcoma, entre otros.
3. HISTIOCITOMA FIBROSO MALIGNO
El término HFM lo introdujeron O´Brien y Stout para tumores de tipo fibroquístico.
Weiss y Enzinger describieron el histiocitoma fibroso maligno mixoide, que comparte varias características con el mixofibrosarcoma, y lo clasificaron en grados, de acuerdo con su histología: bajo grado (predominio mixoide), grado intermedio (mixto: mixoide y celular) y alto grado (predominantemente celular). Sin embargo, en el 2002, la Organización Mundial de la Salud clasificó al histiocitoma fibroso maligno como una entidad y determinó que el tipo mixoide sin factores miogénicos, lipoblásticos y condrogénicos se clasifica como mixofibrosarcoma.
El HFM es el sarcoma de tejidos blandos más común del adulto. La mayor incidencia se sitúa entre la quinta y sexta década de la vida; su presentación resulta inusual antes de los 40 años, con una edad media de 52 años. Presenta cierta predilección por el sexo masculino y se localiza predominantemente en las extremidades. Otros sitios menos comunes son el retroperitoneo, la cabeza y el cuello.
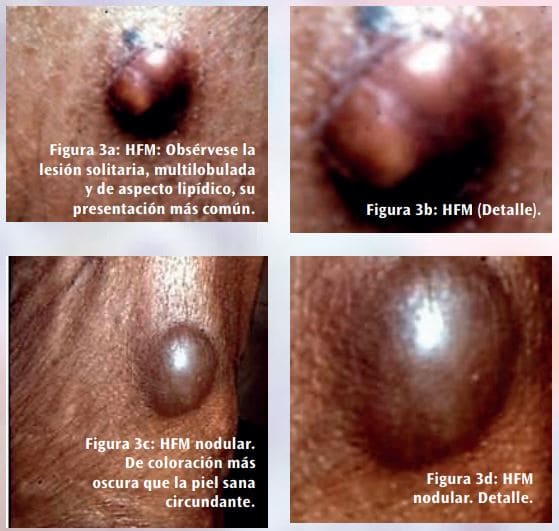
Se presenta en forma multilobulada, superficial (por lo general se origina en la zona subcutánea), aparentemente bien delimitado pero poco encapsulado, de tamaño variable (entre 3 y 38 centímetros, siendo su tamaño más habitual de alrededor de los 6 centímetros), de color gris y con áreas de necrosis y hemorragia de coloración rojiza u oscura. La consistencia es blanda (Figura 3).
Supone del 5 al 10% de los sarcomas de partes blandas. Se deriva de las células mesenquimales primitivas, capaces de diferenciarse hacia fibroblasto, miofibroblasto, histiocitotisular y células intermedias fibrohistiocitarias. El 26% se encuentran en las extremidades, siendo el tumor más frecuente en las personas mayores de 65 años que presentan sarcomas de partes blandas. También hay algunos HFM que aparecen en tejido óseo.
En cuanto a su etiología, continúa siendo desconocida con certeza. La hipótesis histogenética más aceptada es la que sitúa a las células mesenquimales pluripotenciales como precursoras neoplásicas, lo que justifica que en ocasiones coincida temporalmente con otros tumores como el liposarcoma o el fibrosarcoma. También se ha sugerido la posibilidad de que este tumor aparezca, en algunos casos, en áreas previamente radiadas. Como sucede con otros sarcomas, también se ha asociado con las lesiones del síndrome de Dupuytren. En el caso de los localizados en el hueso, se han asociado con algunos procedimientos quirúrgicos y con osteomielitis crónica.
Aproximadamente el 55% de los HMF recidivan y el 48% metastatizan. Los del tipo mixoide son los que tienen mejor pronóstico. En general, son signos de buen pronóstico los subtipos morfológicos estoriforme y mixoide, el menor tamaño del tumor y la ausencia de invasión vascular.
El infiltrado inflamatorio compuesto por linfocitos-T (muy común con otras neoplasias y lesiones), oscurece el diagnóstico del HFM, por lo que se requiere diagnóstico diferencial (generalmente inmunohistoquímico) con infiltrados inflamatorios, pseudotumor inflamatorio, linfoma, leiomiosarcoma inflamatorio y carcinoma de células escamosas. Por otra parte, algunos autores, cuestionan que el HFM sea una entidad tumoral singular y sugieren que es una fase evolutiva del leiomiosarcoma.
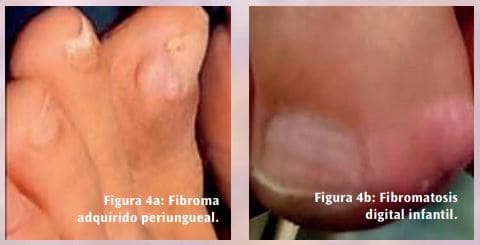
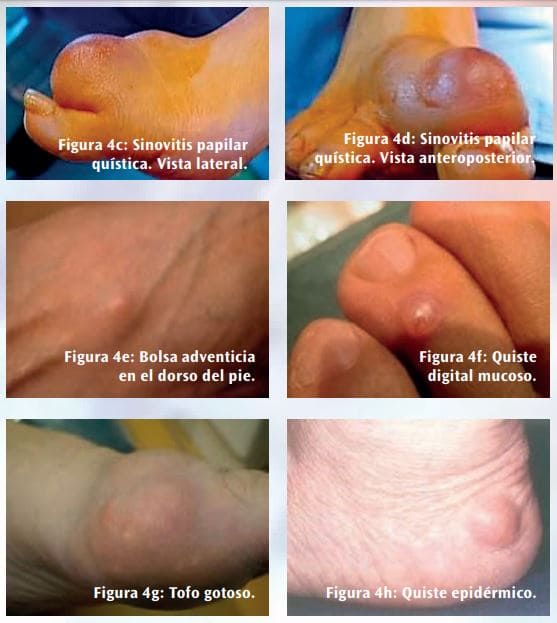
El diagnóstico clínico también es complejo. Se han comunicado casos en los que el diagnóstico inicial de un HFM fue de carcinoma sarcomatoide, fibroxantoma atípico y leiomiosarcoma. Sus características clínicas en algunos de sus estadios evolutivos pueden enmascararlo en lesiones y tumores benignos como el histiocitoma benigno, con fibromas y fibromatosis en sus diversas presentaciones (Figuras 4a y 4b), una sinovitis papilar quística (Figuras 4c y 4d), una bolsa adventicia (Figura 4e), un quiste digital mucoso (Figura 4f), un Tofo gotoso (Figura 4g) y un quiste epidérmico (Figura 4h), entre otros.
DISCUSIÓN
Los sarcomas de partes blandas (SPB) constituyen un grupo heterogéneo de neoplasias originarias de las células mesenquimales. Las células mesenquimales están presentes en todas las regiones anatómicas y, por tanto, los SPB se pueden desarrollar en cualquier parte del organismo humano.
Los diferentes tipos de células mesenquimales desarrollan tumores específicos benignos y malignos. Globalmente las variedades benignas son de 5 a 7 veces más frecuentes que sus equivalentes de SPB. Sin embargo, hay que tener en cuenta que, habitualmente, la gran mayoría de SPB se originan en regiones anatómicas sin ninguna lesión benigna preexistente.
Por otra parte, las alteraciones en la circulación linfática como edemas y linfedemas crónicos, congénitos o secundarios a vaciamientos ganglionares se relacionan con un mayor riesgo a desarrollar SPB in situ. También se originan con mayor prevalencia en las zonas subyacentes a ulceraciones crónicas. Hay descritos varios sistemas de evaluación de los estados evolutivos de los SPB.
El sistema de clasificación más ampliamente aceptado producido conjuntamente por el Comité Mixto Americano de Cáncer (AJCC) y la Unión Internacional contra el Cáncer (UICC) incluye información sobre el grado y la etapa del tumor (tomado de):
TUMOR PRIMARIO (T)
- TX: El tumor primario no puede ser evaluado
- T0: No hay evidencia de tumor primario
- T1: Tumor ≤5 cm en la mayor dimensión
- T1a: Tumor superficial
- T1b: Tumor profundo
- T2: Tumor >5 cm en su mayor dimensión
- T2a: Tumor superfi cial
- T2b: Tumor profundo
GANGLIOS LINFÁTICOS REGIONALES (N)
- NX: Los ganglios linfáticos regionales no pueden ser evaluados
- No: Ninguna metástasis regional de ganglios linfáticos
- N1: Metástasis ganglionar regional
METÁSTASIS A DISTANCIA (M)
- MX: La metástasis a distancia no puede ser evaluada
- M0: No hay metástasis a distancia
- M1: Metástasis a distancia
GRADO HISTOLÓGICO (G):
El sistema de la AJCC usa 4 grados histológicos, mientras que el sistema recomendado en el Reino Unido (FNCLCC) usa 3. Los grados coincidentes son G1 = grado bajo, G2 = grado intermedio y G3 y G4 = grado alto.
- GX: El grado no puede ser evaluado
- G1: Bien diferenciado
- G2: Moderadamente diferenciado
- G3: Pobremente diferenciado
- G4: Pobremente diferenciado o indiferenciado
El agrupamiento de la etapa final es el siguiente:
Etapa I
- 1A: Grado bajo, pequeño, superfi cial o profundo (G1-2, T1a-b, N0, M0).
- 1B: Bajo grado, grande, superfi cial (G1-2, T2a, N0, M0).
Etapa II
- IIA: Bajo grado, grande, profundo (G1-2, T2b, N0, M0).
- IIB: Alto grado, pequeño, superfi cial o profundo (G3- 4, T1a-b, N0, M0).
- IIC: Alto grado, grande, superfi cial (G3-4, T2a, N0, M0).
Etapa III
Alto grado, grande, profundo (G3-4, T2b, N0, M0).
Etapa IV
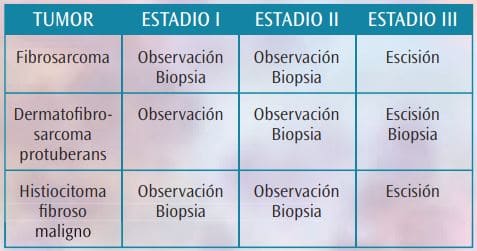
Cualquier metástasis (cualquier G, cualquier T, N1 o M1). De acuerdo con estos estadios, de un modo resumido, proponemos las siguientes pautas de actuación de cara al diagnóstico clínico de los tumores malignos del pie de origen fibroso (Tabla 1).
CONCLUSIONES
- Por todo lo anteriormente expuesto, como primera conclusión, debemos insistir en la importancia de una exhaustiva historia clínica que aproxima al profesional a conocer lo más exactamente posible el momento de aparición de la primera manifestación dermatológica, el tiempo de evolución de la lesión y la existencia o no de factores de riesgo en el pasado o en el momento presente.
- Es fundamental guardar un registro fotográfico de la lesión primitiva, tal como la hemos visto en la primera visita. La evolución de las lesiones aparentemente benignas hacia un SCB, sin ser frecuente, puede suceder. Por otra parte, los SPB adoptan diversas formas de presentación en sus diversas fases evolutivas.
- Mantener una información actualizada acerca de las diversas formas de presentación clínica, epidemiología y etiopatogenia de los SPB ayudará al profesional a realizar un diagnóstico precoz de estas neoplasias.
VALERO José*, SARROCA Nuria**.
* Podólogo y Antropólogo. Especialista en Cirugía Podológica. Doctor por la Universidad de Zaragoza (Sociología). Doctor por la Universidad de Zaragoza (Medicina).
** Graduada en Podología. Master en Investigación en Podología (Universidad Rey Juan Carlos).
Laboratorios Herbitas es una compañía especializada en la fabricación y distribución de productos podológicos y de fisioterapia. Poseemos 30 años de experiencia en el mercado clínico.
Gracias a nuestra experiencia y contacto con los profesionales en biomecánica y cuidado del pie, nuestros productos son diseñados y aprobados por reconocidos profesionales del sector.
Contamos con una de las gamas más amplias del mercado, estando presentes en más de 40 países de Europa, África y América.